Tutorial 08: Analysis Along the Reaction Coordinate¶
Objectives¶
This tutorial will teach:
Extraction of molecular properties from
.outfilesAnalysis of reaction coordinates
Overview¶
Analysis of higher-order properties along a potential energy surface, although powerful, is frequently complicated by the multitude of jobs required. For instance, analyzing the change in atomic charges or aromaticity (as measured by nucleus-independent chemical shift, or NICS) requires a separate job for each desired point, making manual setup and analysis challenging. In contrast, cctk’s ability to automatically extract structures and properties from output files allows for detailed investigations to be carried out with minimal brute-force labor.
For this example, we chose to analyze the carbonyl-ene reaction of acetaldehyde and propylene. This reaction results in a net allylation of a carbonyl compound using inexpensive propylene gas as a surrogate for more costly and promiscuous allylating reagents (like allyl–MgBr), but its utility is hampered by the slow rate of reaction: 26.4 kcal/mol for formaldehyde (ref)) and ~39 kcal/mol for aliphatic aldehydes (independent results). Effective catalysis of the carbonyl–ene reaction, guided by improved understanding of transition state properties, could therefore lead to an effective and powerful synthetic method.
To study this reaction, we chose to investigate:
The dipole moment
Hirshfeld charges of key heavy atoms
NICS(0) of the ring formed by the two reactants
Finding Points Along the Reaction Coordinate¶
The transition state for the reaction was found using conventional techniques (scanning the C1–C5 bond distance)
and confirmed with a frequency calculation (vi = –1336 cm-1)
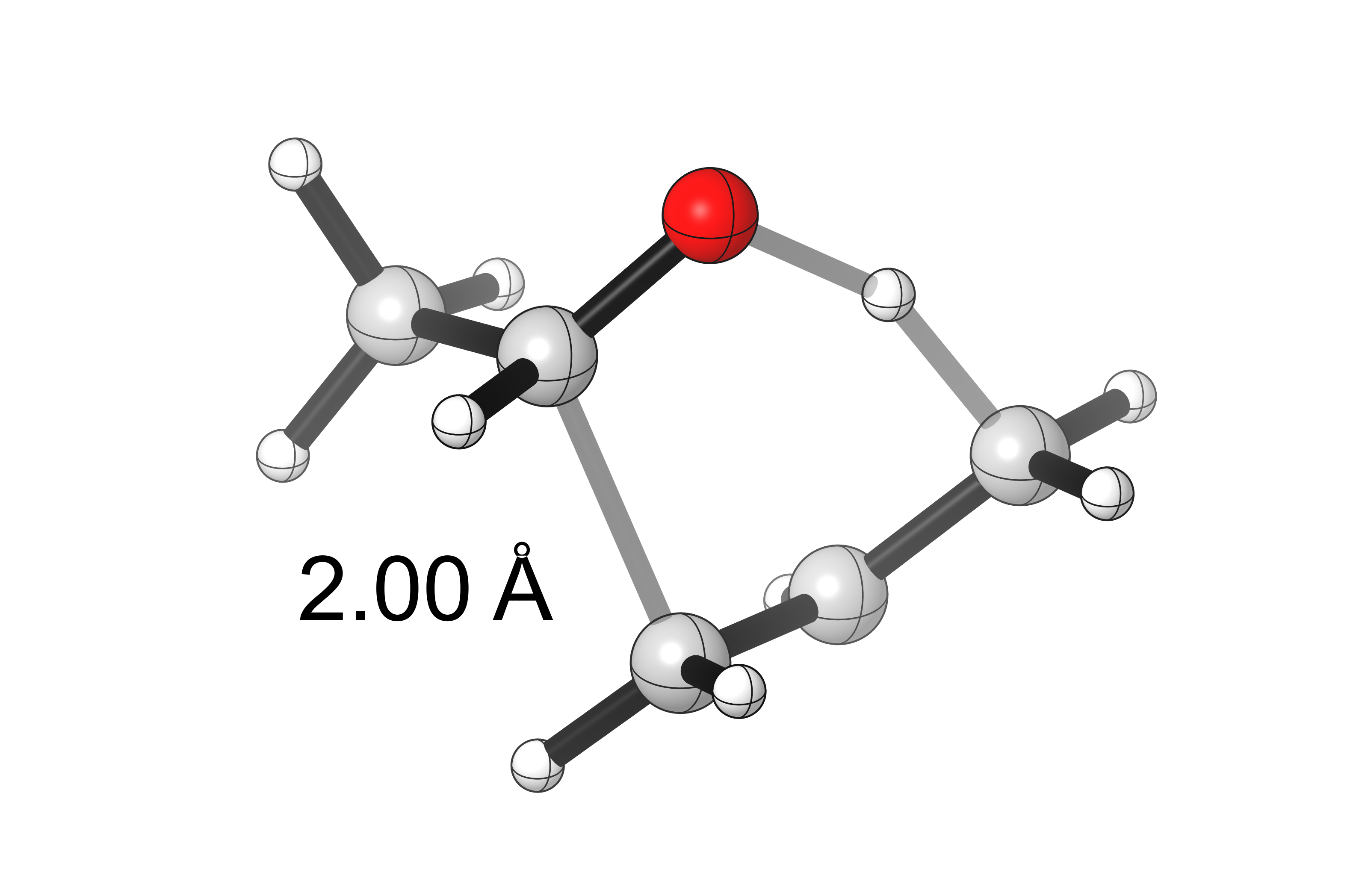
The intrinsic reaction coordinate was followed backwards and forwards for 50 steps using the following input line:
#p irc=(calcfc, forward, maxpoints=50, stepsize=2) m062x/6-31g(d)
This resulted in the generation of two IRC .out files, each containing 51 distinct structures.
IRC calculations are notoriously error-prone; direct scans can be used as a surrogate as needed.
Generating Jobs to Calculate NICS(0)/Hirschfeld Charges¶
The structures from the IRC .out files must first be extracted before new jobs can be written.
To populate a ConformationalEnsemble object with the structures from the IRC, we used the join_ensembles() method:
for filename in glob.iglob(args["filename"], recursive=True):
if re.search("slurm", filename):
continue
output_file = GaussianFile.read_file(filename)
ensembles.append(output_file.molecules)
new_ensemble = ConformationalEnsemble.join_ensembles(ensembles)
The files are then generated (and named after the C1–C8 distance, which decreases monotonically along the IRC).
For NICS calculation, a “ghost atom” must be added at the centroid of the ring
(symbol “Bq”, after the ghost Banquo from Macbeth).
The Hirshfeld population jobs die if ghost atoms are used, so separate files are required (generate_nics.py and generate_pop.py):
for mol in new_ensemble.molecules:
mol.add_atom_at_centroid("Bq", [1, 7, 15, 12, 9, 8])
cc_dist = mol.get_distance(1, 8)
newfile = f"nics_{int(round(cc_dist*1000))}.gjf"
GaussianFile.write_molecule_to_file(newfile, mol, "#p nmr m062x/6-31g(d)", None)
print(f"generating {newfile}...")
The jobs should then be submitted – they should each take no more than a few minutes to run.
Analysis¶
Since the information in the .out files is not automatically extracted by GaussianFile.read_file(),
more sophisticated methods for automated parameter extraction must be employed.
parse_gaussian.py (imported as parse) contains several awk-like methods for locating blocks of text in .out files:
find_parameter() is useful for locating key values on a given line, while search_for_block() extracts larger blocks of text.
(Note that return_lines=True must be set on GaussianFile.read_file() to generate the requisite lines variable).
A part of graph.py is shown below:
for filename in sorted(glob.glob(filenames, recursive=True)):
if re.search("slurm", filename):
continue
(output_file, lines) = GaussianFile.read_file(filename, return_lines=True)
dist = int(round(output_file.get_molecule().get_distance(1, 8) * 1000))
energies[dist] = output_file.energies[-1]
try:
nics[dist] = -1 * parse.find_parameter(lines, "17 Bq Isotropic", 8, 4)[0]
except:
pass
try:
dipole_line = parse.search_for_block(lines, "Dipole", "Quadrupole")
fields = re.split(" +", dipole_line)
fields = list(filter(None, fields))
dipole[dist] = float(fields[-1])
except:
pass
try:
C1_charge[dist] = parse.find_parameter(lines, " 1 C", 8, 2)[-1]
O7_charge[dist] = parse.find_parameter(lines, " 7 O", 8, 2)[-1]
C8_charge[dist] = parse.find_parameter(lines, " 8 C", 8, 2)[-1]
C9_charge[dist] = parse.find_parameter(lines, " 9 C", 8, 2)[-1]
C12_charge[dist] = parse.find_parameter(lines, " 12 C", 8, 2)[-1]
except:
pass
With these values in hand, we can generate graphs showing change along the IRC:
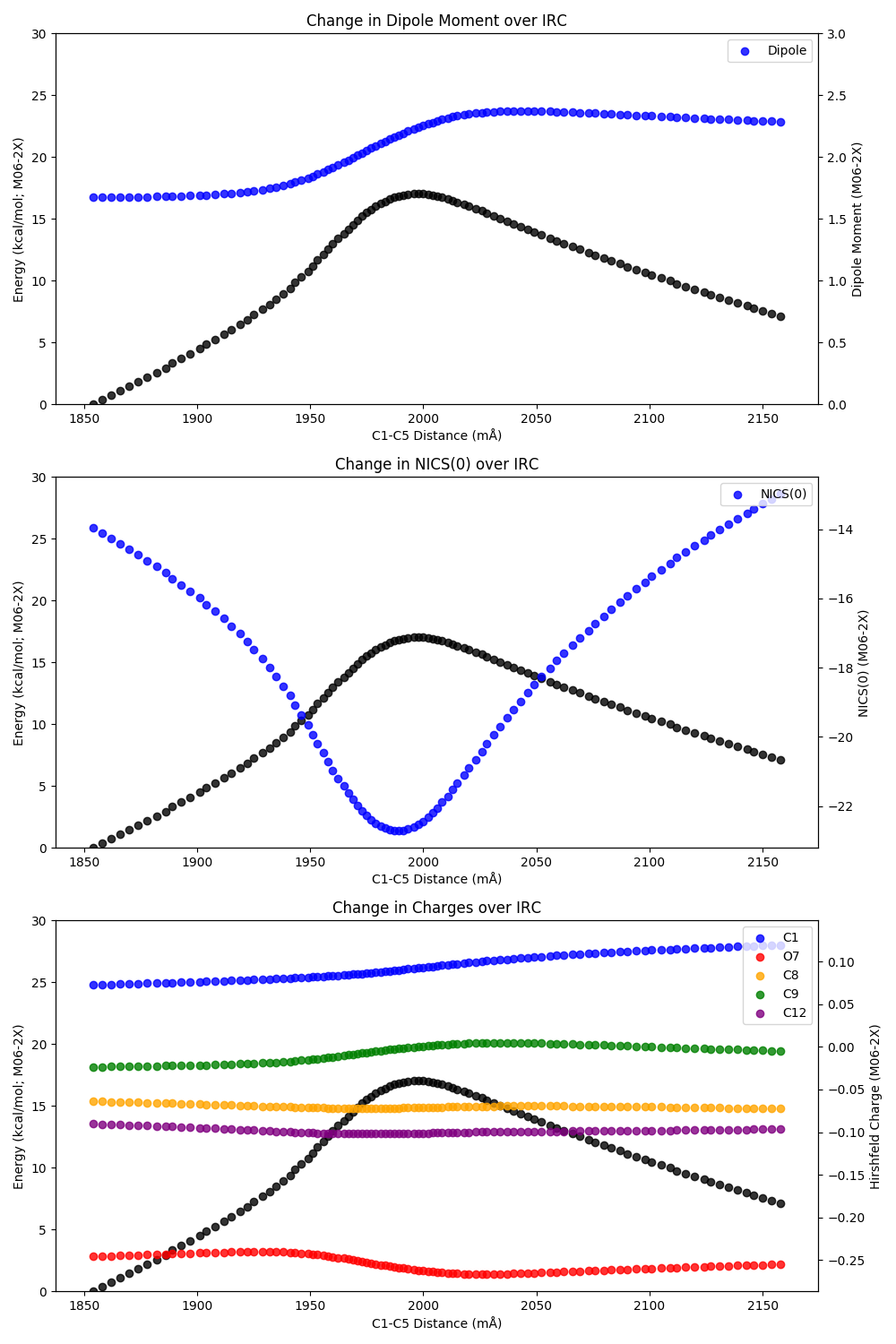
As shown by these images, the reaction proceeds without significant buildup of positive or negative charge and with minimal change in the overall dipole moment, indicating solvent polarity is unlikely to drastically raise/lower the rate of reaction. However, the drastic drop in NICS(0) around the transition state indicates that a true aromatic transition state is present, consistent with Schleyer’s findings (see Section 3.8.5). Accordingly, pi–pi stacking or other aromatic-stabilizing interactions might be a fruitful avenue for catalyst design to accelerate this reaction.
A more in-depth study might analyze the role of open-shell or multiconfigurational species (like in this paper), as well as investigating how Lewis acid catalysts like Et2AlCl change the above properties.