Tutorial 06: Performing a Conformational Search¶
Objectives¶
This tutorial will teach:
Automated analysis and resubmission of completed jobs.
Creation of multi-step cctk projects.
Overview¶
Predicting the conformation of short peptides in solution is a challenging and unsolved problem. This “conformational heterogeneity” is both theoretically interesting and of practical import for the design and study of catalytically active oligopeptides (e.g., Scott Miller’s catalysts). In particular, the inclusion of unnatural amino acids in peptides could allow for stable conformations completely unlike those obtained using canonical amino acids.
One such unnatural amino acid is difluoroalanine, which possesses a difluoromethyl group. The difluoromethyl group has recently been implicated as a hydrogen bond donor, albeit with distinct properties from classic hydrogen bond donors like hydroxyl or amino groups.
To examine potential C–H and N–H hydrogen bonding interactions involving difluoroalanine, we chose to study the 31-atom dipeptide Ac–F2Ala–F2Ala–OMe.
Generating Conformations to Search¶
Although in principle there are a practically infinite number of distinct structures that can be generated from 31 atoms,
in practice most of low-energy conformational space can be sampled
by selecting a few key rotatable bonds and letting Gaussian’s opt keyword do the rest.
- For the purposes of this study, we chose to study four key dihedral angles:
Rotating around F2Ala #1’s alpha carbon with respect to the amide (C3–C5–C8–O6)
Rotating around F2Ala #2’s alpha carbon with respect to the amide (H12–C11–C14–O16)
Rotating the difluoromethyl group in F2Ala #1 (N1–C3–C5–H7)
Rotating the difluoromethyl group in F2Ala #2 (N9–C11–C13–H15)
- We also chose to sample:
cis/trans isomerism in F2Ala #1 (O27–C26–N1–H2)
cis/trans isomerism in F2Ala #2 (O8–C6–N9–H10)
Methyl ester conformation (O16–C14–O17–C18)
Each of the dihedral angles in the first list was set to 0, 120, and 240 degrees, while each of the dihedral angles in the second list was set to 0 and 180 degrees (648 conformations in total).
To get our starting Molecule object, we read from an .xyz file. Since .xyz files don’t contain connectivity information,
we have to generate the bonds automatically:
output_file = XYZFile.read_file('Ac-F2Ala-F2Ala-OMe.xyz')
output_file.molecule.assign_connectivity()
The actual heavy lifting is done by the following code, which creates copies of the reference structure with the selected
dihedral angles set to new values. To avoid nesting for loops, we’ll use recursion to run this block once per angle:
new_structures = [None] * (len(structures) * len(thetas))
current_idx = 0
for structure in structures:
for theta in thetas:
new_structures[current_idx] = copy.deepcopy(structure.set_dihedral(*angles[idx], theta, check_result=False))
current_idx += 1
Finally, the script writes the resultant structures to .gjf files. The complete script (generate_conformers.py) is shown below:
import numpy as np
import copy
from cctk import XYZFile, ConformationalEnsemble, GaussianFile
#### Usage: ``python generate_conformers.py``
#### This script takes an input ``.xyz`` file and outputs ~600 different conformations.
#### Corin Wagen and Eugene Kwan, 2019
output_file = XYZFile.read_file('Ac-F2Ala-F2Ala-OMe.xyz')
output_file.molecule.assign_connectivity()
#### here we define the different choices for each angle.
angles = range(0, 360, 120)
bin_angles = range(0, 360, 180)
#### here we define all the rotatable bonds, and the bonds with two conformers (like amides)
to_rotate = [[1, 3, 5, 7], [9, 11, 13, 15], [5, 3, 6, 8], [12, 11, 14, 16]]
to_bin_rotate = [[27, 26, 1, 2], [8, 6, 9, 10], [16, 14, 17, 18]]
#### now to employ some recursion...
def rotate_angles_one_by_one (idx, angles, thetas, structures):
"""
This script takes a set of structures, and outputs a (longer) set of structures where the given bond has been rotated.
Args:
idx (int): the current position in ``angles``
angles (list of 4-element lists): the list of angles to recurse through and adjust
thetas (list of float): the list of dihedral angles to set each bond to
structures (list of cctk.Molecule): the current list of structures
Returns:
list of cctk.Molecule objects (with len(thetas) * len(structures) elements)
"""
if idx >= len(angles):
return structures
else:
new_structures = [None] * (len(structures) * len(thetas))
current_idx = 0
for structure in structures:
for theta in thetas:
new_structures[current_idx] = copy.deepcopy(structure.set_dihedral(*angles[idx], theta, check_result=False))
current_idx += 1
mols = rotate_angles_one_by_one(0, to_rotate, angles, [output_file.molecule])
mols = rotate_angles_one_by_one(0, to_bin_rotate, bin_angles, mols)
for idx, molecule in enumerate(mols):
try:
molecule.check_for_conflicts()
GaussianFile.write_molecule_to_file(f"conformer_{idx:05d}.gjf", molecule, "#p opt pm7", None)
except:
pass
The resultant Gaussian jobs (optimizations using the quick semiempirical pm7 method) should converge quickly.
Analyzing and Resubmitting¶
After running all the jobs (which should take a few hours), we need to analyze the results.
We do this by reading in all the files and adding the molecules to a ConformationalEnsemble object (along with their energies):
for filename in sorted(glob.glob(filenames, recursive=True)):
if re.search("slurm", filename):
continue
try:
output_file = GaussianFile.read_file(filename)
if len(output_file.energies) > 0:
mol = output_file.get_molecule()
ensemble.add_molecule(mol, energy=output_file.energies[-1]*627.509)
except:
print(f"skipping f{filename} due to error...")
The next step is to eliminate redundant conformers (since there aren’t 648 distinct low-energy conformations of this peptide, many of the jobs will have converged to the same structure).
By using a similarity threshold of 0.6, 166 distinct structures can be obtained:
print(f"{len(ensemble.molecules)} conformers before elimination of redundant")
ensemble.eliminate_redundant(cutoff=0.6)
print(f"{len(ensemble.molecules)} conformers after elimination of redundant")
Finally, all the conformers within 10 kcal/mol of the pm7 global minimum are resubmitted using DFT and implicit solvation:
best_confs = ensemble.get_within_cutoff(cutoff=10)
for idx, molecule in enumerate(best_confs):
GaussianFile.write_molecule_to_file(f"conformer_v2_{idx:03d}.gjf", molecule, "#p opt freq=noraman m062x/6-31g(d) scrf=(smd,solvent=diethylether)", None)
The script also outputs the energy and key dihedral angles for all 166 distinct conformers. The full script (extract_unique.py) is shown below:
import sys, re, glob
import numpy as np
from cctk import GaussianFile, Molecule, ConformationalEnsemble
#### This is a script to extract the lowest-energy unique conformers and resubmit them at a higher level of theory.
#### By default, this file will create new ``.gjf`` files for any conformer within 10 kcal/mol of the lowest-energy conformer.
#### Usage: ``python extract_unique.py "path/to/output/*.out"``
#### NOTE: It's crucial to wrap the wildcard-containing path in quotes!
#### Corin Wagen and Eugene Kwan, 2019
filenames = sys.argv[1]
info = []
text_width = 70
to_rotate = [[1, 3, 5, 7], [9, 11, 13, 15], [5, 3, 6, 8], [12, 11, 14, 16]]
ensemble = ConformationalEnsemble()
for filename in sorted(glob.glob(filenames, recursive=True)):
if re.search("slurm", filename):
continue
try:
output_file = GaussianFile.read_file(filename)
if len(output_file.energies) > 0:
mol = output_file.get_molecule()
ensemble.add_molecule(mol, energy=output_file.energies[-1]*627.509)
except:
print(f"skipping f{filename} due to error...")
print(f"{len(ensemble.molecules)} conformers before elimination of redundant")
ensemble.eliminate_redundant(cutoff=0.6)
print(f"{len(ensemble.molecules)} conformers after elimination of redundant")
best_confs = ensemble.get_within_cutoff(cutoff=10)
for idx, molecule in enumerate(best_confs):
GaussianFile.write_molecule_to_file(f"conformer_v2_{idx:03d}.gjf", molecule, "#p opt freq=noraman m062x/6-31g(d) scrf=(smd,solvent=diethylether)", None)
for idx, molecule in enumerate(list(ensemble.molecules[np.argsort(ensemble.energies)])):
items = [idx+1, np.sort(ensemble.energies)[idx]]
for atoms in to_rotate:
items.append(molecule.get_dihedral(*atoms))
if idx==0:
print("Molecule Energy D" + str(" D".join(str(a) for a in to_rotate)))
print(str(" ".join(str(x)[0:8] for x in items)))
Final Analysis and Visualization¶
The high-level results can be subjected to the same elimination of redundant conformers, which yields 7 final structures. The lowest-energy structure is the linear form, but several other structures contain close N–H to C=O contacts. None of the structures studied appears to contain a close C–H to C=O contact, indicating that difluoromethyl hydrogen bonding is not significant in this structure. Instead, the difluoromethyl groups appear to be oriented so as to minimize the overall molecular dipole.
Although we chose input structures with a 10 kcal/mol difference in energies, the output files are all within roughly 5 kcal/mol (∆G). This may be due to increased shielding of dipole/dipole interactions due to implicit solvation.
When run, the analysis script yields the following output:
$ python analyze_final.py "output/*v2*.out"
12 conformers before elimination of redundant
7 conformers after elimination of redundant
writing final conformers to disk as ``conformer_final_xx.gjf``...
Molecule Energy D[1, 3, 5, 7] D[9, 11, 13, 15] D[5, 3, 6, 8] D[12, 11, 14, 16]
0 00.000 066.23 313.11 113.17 235.38
1 00.140 310.90 308.97 121.98 048.58
2 00.616 172.56 309.14 359.35 047.92
3 00.717 064.58 186.38 019.56 241.59
4 01.803 294.45 183.31 019.67 074.00
5 04.466 174.92 302.01 000.30 041.05
6 05.245 060.83 309.23 039.52 240.81
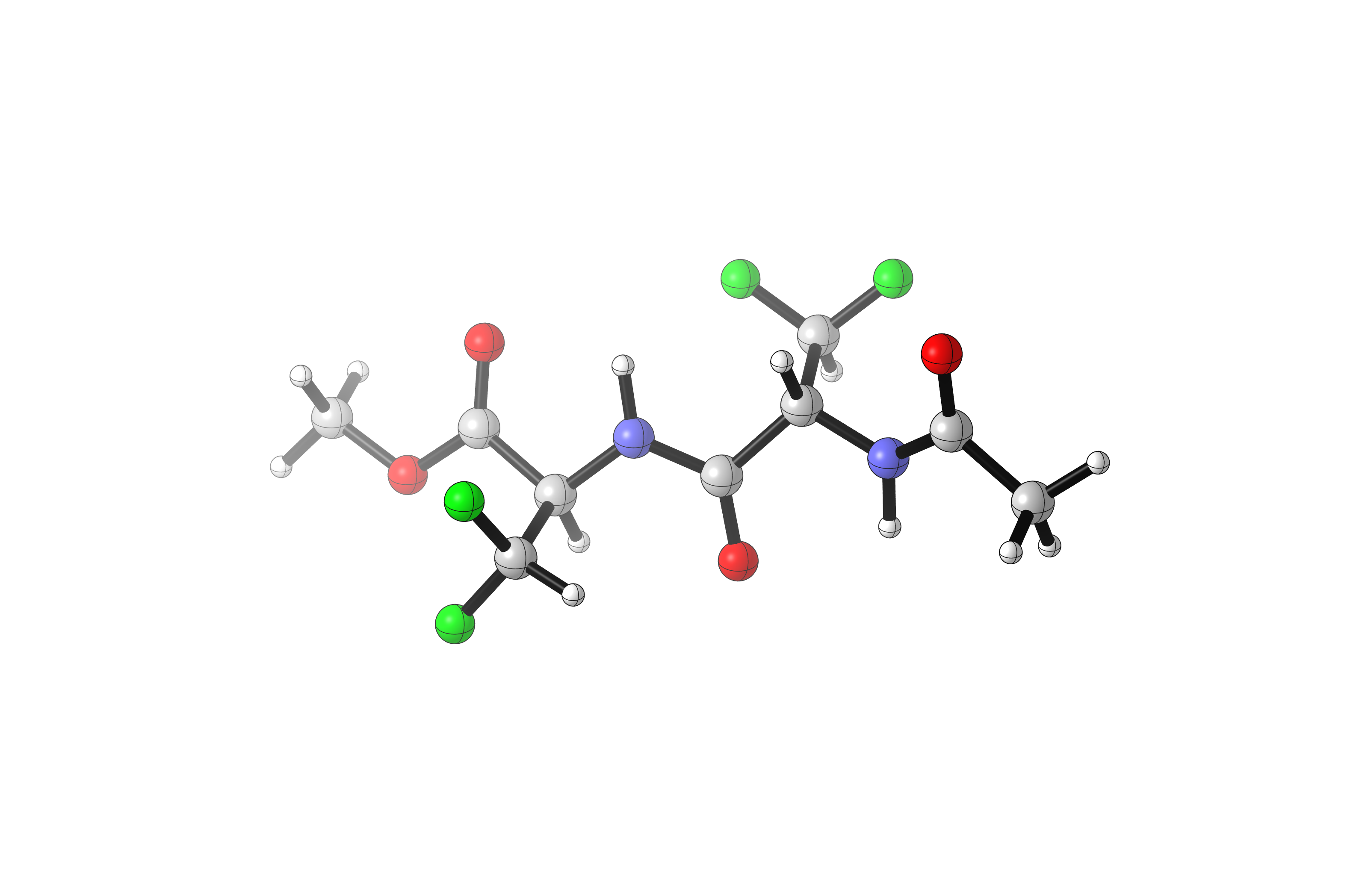
The lowest energy structure (conformer_final_00.gjf) is pictured here—but three other conformations are nearly isoenergetic, so clearly there are many structures which could be relevant to reactivity through a Curtin–Hammett-type scenario.
The full analysis script (analyze_final.py) is as follows:
import sys, re, glob
import numpy as np
from cctk import GaussianFile, Molecule, ConformationalEnsemble
#### This is a script to monitor the final output of the conformational search.
#### This script will print the dihedral angles of the final structures, and output gjf files for each of the final scripts.
#### Usage: ``python analyze_final.py "path/to/output/*.out"``
#### NOTE: It's crucial to wrap the wildcard-containing path in quotes!
#### Corin Wagen and Eugene Kwan, 2019
filenames = sys.argv[1]
info = []
text_width = 70
to_rotate = [[1, 3, 5, 7], [9, 11, 13, 15], [5, 3, 6, 8], [12, 11, 14, 16]]
ensemble = ConformationalEnsemble()
for filename in sorted(glob.glob(filenames, recursive=True)):
if re.search("slurm", filename):
continue
try:
output_file = GaussianFile.read_file(filename)
if len(output_file.energies) > 0:
mol = output_file.get_molecule()
ensemble.add_molecule(mol, energy=output_file.gibbs_free_energy*627.509)
except:
print(f"skipping f{filename} due to error...")
print(f"{len(ensemble.molecules)} conformers before elimination of redundant")
ensemble.eliminate_redundant(cutoff=0.6)
print(f"{len(ensemble.molecules)} conformers after elimination of redundant")
print("writing final conformers to disk as ``conformer_final_xx.gjf``...")
for idx, molecule in enumerate(list(ensemble.molecules[np.argsort(ensemble.energies)])):
items = [idx, f"{np.sort(ensemble.energies)[idx] - np.min(ensemble.energies):06.3f}"]
GaussianFile.write_molecule_to_file(f"conformer_final_{idx:02d}.gjf", molecule, "#p opt freq=noraman m062x/6-31g(d) scrf=(smd,solvent=diethylether)", None)
for atoms in to_rotate:
items.append(f"{molecule.get_dihedral(*atoms):0>6.2f}")
if idx==0:
print("Molecule Energy D" + str(" D".join(str(a) for a in to_rotate)))
print(str(" ".join(str(x)[0:10] for x in items)))