Tutorial 04: Using Groups¶
Objectives¶
This tutorial will teach:
Creating and manipulating
Groupobjects.Combining multiple jobs via the Link1 command.
Overview¶
Substituent effects and linear free-energy relationships are an invaluable tool in physical organic chemistry. More recently, linear free-energy relationships have become important in the design of new ligands for organometallic chemistry. This approach, wherein key ligand properties are parameterized and used to model reaction outcomes through multilinear regression, has been popularized by Matt Sigman and others for reaction optimization.
Key to this process is the choice of appropriate and informative ligand parameters. One such parameter, termed the Tolman electronic parameter (or ν), is the A1 carbonyl stretching frequency of L–Ni(CO)3 complexes. This stretching vibration is proportional to the degree of Ni(d) to CO(π*) backbonding: more π-acidic ligands compete with CO and reduce the backbonding, thereby shifting the vibration to higher frequencies. (For an excellent review by Tolman, see Chem Rev, 1977, 77, 313–348).
In this tutorial, we will investigate the computed effect of para-substituted triaryl phosphines on this stretching frequency,
using cctk’s Group object to rapidly generate new ligands.
Generating Hammett Series¶
As a starting point, Ph3P–Ni(CO)3 was optimized at the B3LYP-D3BJ/6-31G(d)-SDD(Ni) level of theory. The CO stretching frequency was found to be 2082.1 cm-1.

The para hydrogen atoms on the phenyl rings are numbered 19, 30, and 41 (determined using GaussView).
We can now start to construct our file:
import sys, os, argparse, glob, re, copy
import numpy as np
from cctk import GaussianFile, Molecule, Group, Ensemble
from cctk.load_groups import load_group
parser = argparse.ArgumentParser(prog="hammett_swap.py")
parser.add_argument("filename")
args = vars(parser.parse_args(sys.argv[1:]))
assert args["filename"], "Can't resubmit files without a filename!"
#### read in output file
output_file = GaussianFile.read_file(args["filename"])
#### read in genecp footer
footer = ""
with open('footer', 'r') as file:
footer = file.read()
footer simply contains the genecp basis set definition:
-C -H -N -O -F -S -P -Cl 0
6-31G(d)
****
-Ni
SDD
****
-Ni
SDD
Next, we can define the groups we’re interested in studying:
#### define groups and atoms
groups = ["NMe2", "OMe", "Me", "CF3", "CO2Me", "CN", "NO2", "F", "Cl", "SF5"]
p_atoms = [19, 30, 41]
ensemble = Ensemble()
headers = []
args = []
These groups are all preloaded in cctk, so we don’t need to use any additional input files to define them (for a full list of all available groups, see the Groups documentation).
Finally, we do the actual work of adding the groups:
for group_name in groups:
print(f"adding {group_name}")
mol = copy.deepcopy(output_file.get_molecule().assign_connectivity())
group = load_group(group_name)
for atom in p_atoms:
print(f" adding to atom {atom}")
mol = Group.add_group_to_molecule(mol, group, atom)
ensemble.add_molecule(mol)
headers.append(output_file.header)
args.append({"footer": footer})
We first make a copy of the Molecule object (so we can edit it many times), and load the desired group.
Then, we add the group to every phenyl ring using add_group_to_molecule (this step is relatively slow due to dihedral angle optimization).
Finally, we add the molecule to our Ensemble, and add the header and footer to lists.
We could easily submit each of these as a separate optimization, but to save space it’s sometimes desireable to use Gaussian’s Link1 command to merge multiple disparate jobs into one file.
This can be done here through the write_ensemble_to_file() command:
#### write everything to the same file with Link1
output_file.write_ensemble_to_file("hammett_NiCO3L.gjf", ensemble, headers, args)
The entire script (generate_series.py) is shown below:
import sys, os, argparse, glob, re, copy
import numpy as np
from cctk import GaussianFile, Molecule, Group, Ensemble
from cctk.load_groups import load_group
parser = argparse.ArgumentParser(prog="hammett_swap.py")
parser.add_argument("filename")
args = vars(parser.parse_args(sys.argv[1:]))
assert args["filename"], "Can't resubmit files without a filename!"
#### read in output file
output_file = GaussianFile.read_file(args["filename"])
#### read in genecp footer
footer = ""
with open('footer', 'r') as file:
footer = file.read()
#### define groups and atoms
groups = ["NMe2", "OMe", "Me", "CF3", "CO2Me", "CN", "NO2", "F", "Cl", "SF5"]
p_atoms = [19, 30, 41]
ensemble = Ensemble()
headers = []
args = []
for group_name in groups:
print(f"adding {group_name}")
mol = copy.deepcopy(output_file.get_molecule().assign_connectivity())
group = load_group(group_name)
for atom in p_atoms:
print(f" adding to atom {atom}")
mol = Group.add_group_to_molecule(mol, group, atom)
ensemble.add_molecule(mol)
headers.append(output_file.header)
args.append({"footer": footer})
#### write everything to the same file with Link1
output_file.write_ensemble_to_file("hammett_NiCO3L.gjf", ensemble, headers, args)
Analysis¶
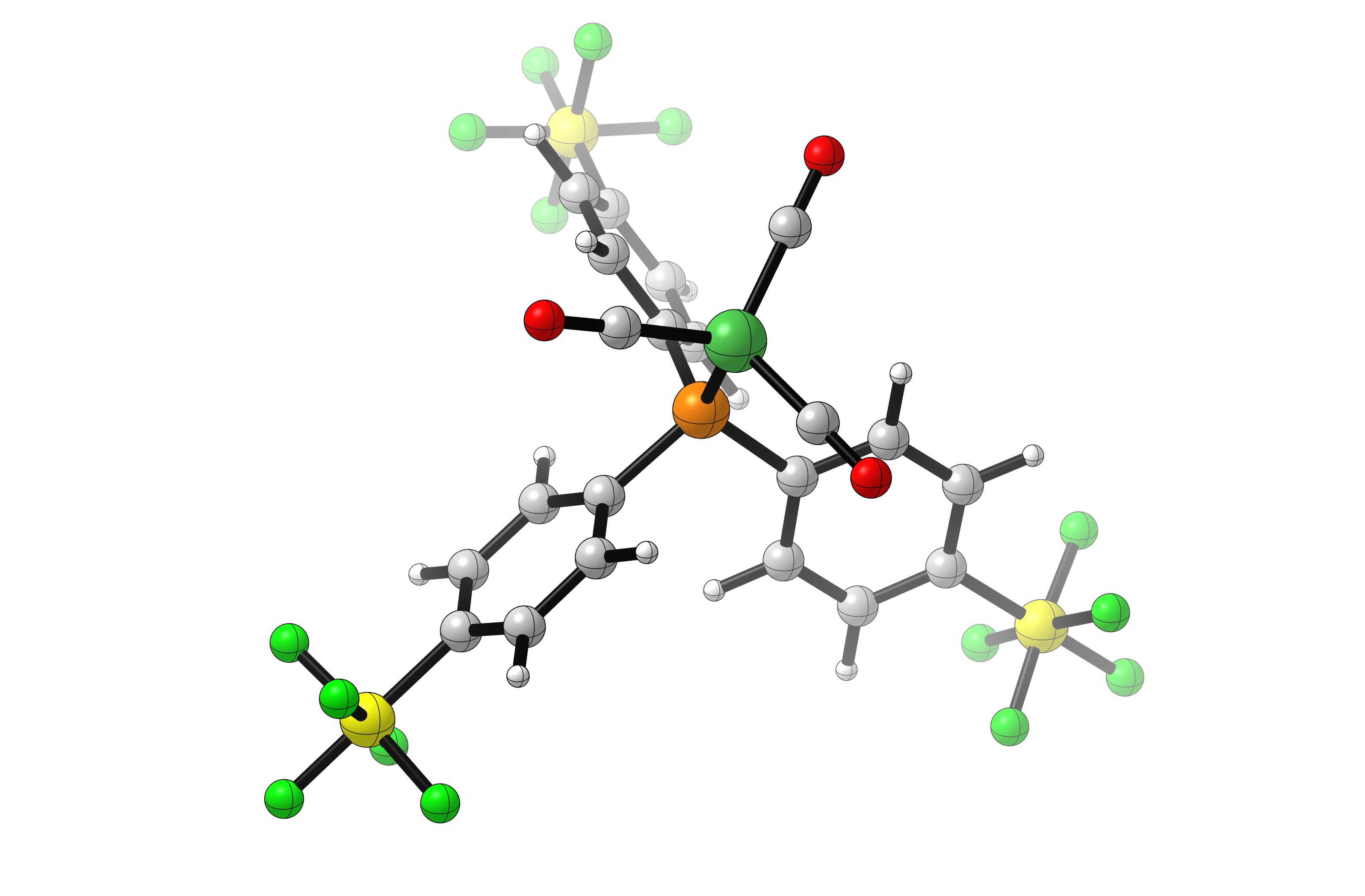
The above job takes about 12 hours to run. Visual analysis of the final structures confirms that group substitution did in fact produce the desired structures (shown here for SF5):
The IR vibrations of each structure can be analyzed in GaussView and the pertinent frequencies extracted. Plotting ν against σp (Chem Rev, 1991, 91, 165–196) gives a clear linear relationship, indicating that this metric does detect changes in ligand electronic structure:
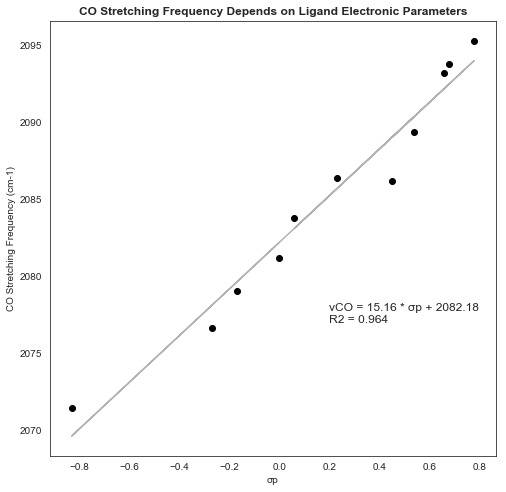
The calculated “Hammett slope” is about 30% steeper than the slope derived from Tolman’s experimental values, implying that this combination of functional and basis set might not be optimal:

Further studies might examine the effect of different computational methods on these computed frequencies, as well as studying the effect of geometric perturbations to the ligand framework.