Tutorial 02: Generating New Conformations¶
Objectives¶
This tutorial will teach:
Manipulation of
MoleculeandEnsembleobjects.Elimination of redundant conformers and automatic checking for steric clashes.
Adjustment of molecular properties.
Overview¶
Generating new conformations for complex molecules can frequently be time-consuming, particularly when one desires to look at multiple rotatable bonds. For butane, there are only three minima about the C2–C3 bond, but looking at the equivalent minima for octane results in 35=243 conformations: far too many to generate manually! Scripting the creation of new conformers with cctk can thus be a powerful time-saving tool.
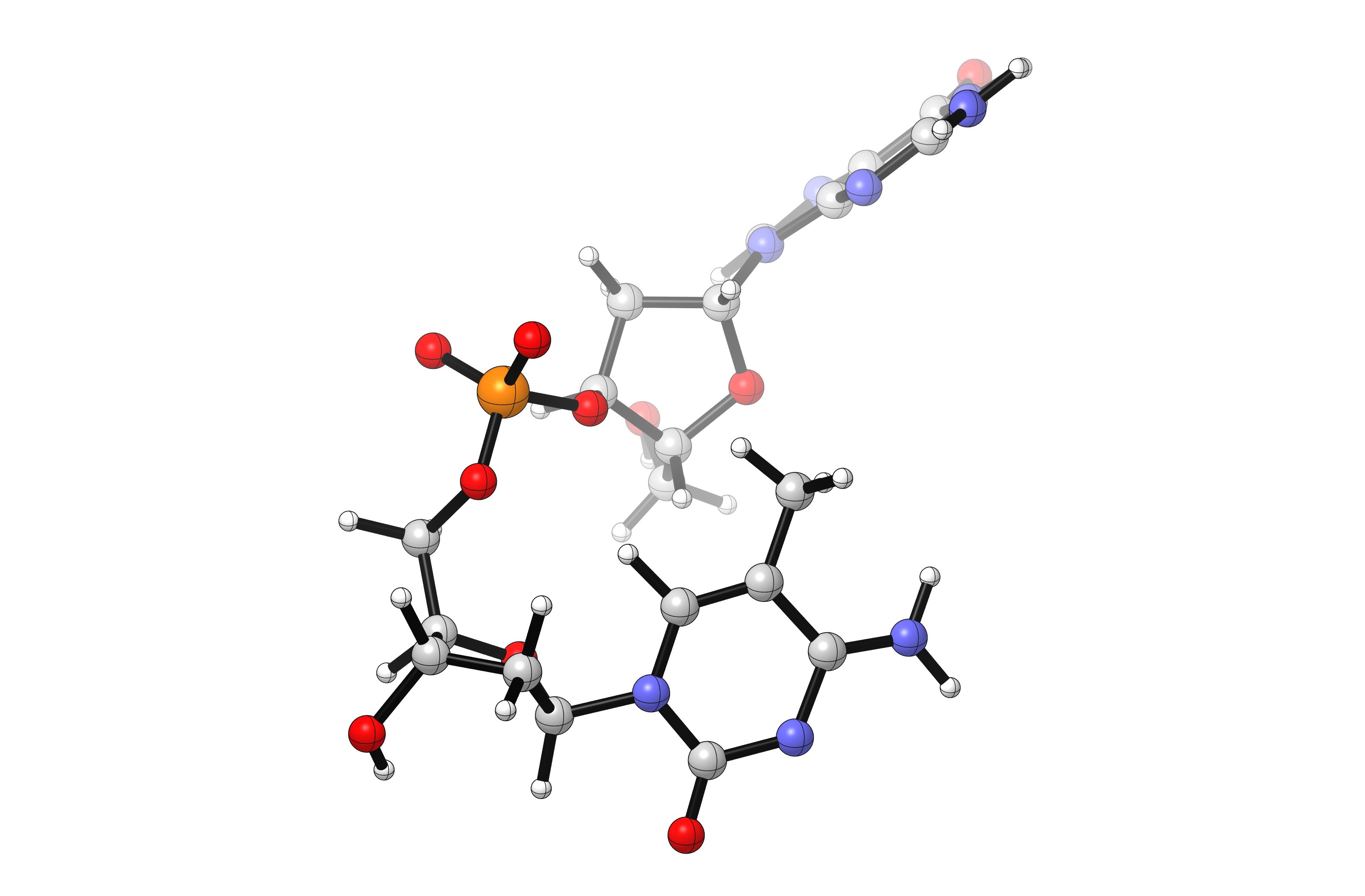
For this tutorial, we will study the N5-methylated CpG dinucleotide (N5-methylation of cytosine is a common repressive epigenetic marker). Although there are many potential conformers of this large molecule, we will focus on rotation about the phosphate group. One rotamer is shown below:
Generating Conformers¶
Create a new file called generate_conformers.py, and import cctk. Next, read in the desired .xyz file and assign the connectivity automatically:
from cctk import XYZFile, ConformationalEnsemble, GaussianFile
output_file = XYZFile.read_file("CpG.xyz")
output_file.molecule.assign_connectivity()
We next want to create a ConformationalEnsemble object to hold our new structures, and define the angles we’ll be looking at:
ensemble = ConformationalEnsemble(name='cpg conformers')
angles = [0, 60, 120, 180, 240, 300]
Now, we want to generate our conformers. New rotamers about the P–O bonds can be generated through the use of molecule.set_dihedral(), and the resultant molecule can be added to the ensemble:
for x in angles:
for y in angles:
output_file.molecule.set_dihedral(1, 7, 6, 8, x)
output_file.molecule.set_dihedral(23, 24, 25, 1, y)
ensemble.add_molecule(output_file.molecule)
ensemble now contains all 62=36 of our desired conformers!
We may wish to check that our conformers are actually distinct, which can be done using ConformationalEnsemble.eliminate_redundant():
old_num = len(ensemble.molecules)
ensemble = ensemble.eliminate_redundant()
new_num = len(ensemble.molecules)
print(f"originally {old_num} conformers, but after eliminating redundant there are {new_num}!")
Creating Input Files¶
After elimination of redundant conformers, the resultant molecules are checked for conflicts (using covalent radii) and then written to .gjf files:
count = 0
for molecule in ensemble.molecules:
x = int(round(molecule.get_dihedral(1, 7, 6, 8)))
y = int(round(molecule.get_dihedral(23, 24, 25, 1)))
try:
molecule.check_for_conflicts()
GaussianFile.write_molecule_to_file(f"conformers/CpG_{x}_{y}.gjf", molecule, "#p opt b3lyp/6-31g(d)", None)
count += 1
except ValueError as e:
print(f"x={x} y={y} no good - {e}")
print(f"wrote {count} molecules to files")
When we run the code, we see that one combination of dihedral angles indeed gives us a steric clash, which would likely crash Gaussian if run:
$ mkdir conformers
$ python generate_conformers.py
originally 36 conformers, but after eliminating redundant there are 36!
x=300 y=60 no good - atoms 8 and 20 are too close - distance 0.4463381332341808 A!
wrote 35 molecules to files
$ ls conformers/
CpG_120_120.gjf CpG_120_300.gjf CpG_180_120.gjf CpG_180_300.gjf CpG_240_120.gjf CpG_240_300.gjf CpG_300_120.gjf CpG_300_300.gjf CpG_360_180.gjf CpG_360_360.gjf CpG_60_120.gjf CpG_60_300.gjf
CpG_120_180.gjf CpG_120_360.gjf CpG_180_180.gjf CpG_180_360.gjf CpG_240_180.gjf CpG_240_360.gjf CpG_300_180.gjf CpG_300_360.gjf CpG_360_240.gjf CpG_360_60.gjf CpG_60_180.gjf CpG_60_60.gjf
CpG_120_240.gjf CpG_120_60.gjf CpG_180_240.gjf CpG_180_60.gjf CpG_240_240.gjf CpG_240_60.gjf CpG_300_240.gjf CpG_360_120.gjf CpG_360_300.gjf CpG_60_0.gjf CpG_60_240.gjf
$ ls conformers/ | wc -l
35
If we try adding purposefully similar angles, we can see that eliminate_redundant works as intended:
angles = [0, 60, 120, 180, 240, 241, 300] # 36 output files written, not 49!
The final script looks like this:
from cctk import XYZFile, ConformationalEnsemble, GaussianFile
#### This file generates a bunch of different phosphate-based rotamers for the given CpG dinucleotide.
#### Usage: ``python generate_conformers.py``
output_file = XYZFile.read_file("CpG.xyz")
output_file.molecule.assign_connectivity()
ensemble = ConformationalEnsemble(name='cpg conformers')
angles = [0, 60, 120, 180, 240, 241, 300]
for x in angles:
for y in angles:
output_file.molecule.set_dihedral(1, 7, 6, 8, x)
output_file.molecule.set_dihedral(23, 24, 25, 1, y)
ensemble.add_molecule(output_file.molecule)
old_num = len(ensemble.molecules)
ensemble = ensemble.eliminate_redundant()
new_num = len(ensemble.molecules)
print(f"originally {old_num} conformers, but after eliminating redundant there are {new_num}!")
count = 0
for molecule in ensemble.molecules:
x = int(round(molecule.get_dihedral(1, 7, 6, 8)))
y = int(round(molecule.get_dihedral(23, 24, 25, 1)))
try:
molecule.check_for_conflicts()
GaussianFile.write_molecule_to_file(f"conformers/CpG_{x}_{y}.gjf", molecule, "#p opt b3lyp/6-31g(d)", None)
count += 1
except ValueError as e:
print(f"x={x} y={y} no good - {e}")
print(f"wrote {count} molecules to files")
The output files can be submitted and the resultant energies compared to determine the ground-state conformational distribution.